The Ising model is an idealized statistical mechanics model of ferromagnetism—the general mechanism that explains how materials can become permanent magnets.
An Ising model represents atoms as variables that can occupy just two spin states, +1 and -1, which are arranged in a graph (usually a simple lattice) where every variable interacts only with its nearest neighbors.
Let’s start by imagining a 1-D Ising model system, so we just have a line of some finite amount of atoms N, labeled start to end as 1 to N. The system is analogous to a mathematical bracelet, where N+1 will “curve” back to the beginning. In other words, even though there are only N atoms, N+1 is actually defined in the system—it’s the same as atom 1. N+2 is then the same as 2, and so on (so generally speaking, kN+x is the same as x, where k is an integer).
The equation for the Hamiltonian H of this system (if you don’t know what that is yet, just think of it as the equation for the total energy) can be represented like this:
Where atoms are represented as S and their subscript denotes the position. Another way of writing the first sum would be:
Notice that every atom is represented in the sum exactly twice (remember, S_N+1 is the same as S_1), so that every relevant interaction is represented–we earlier required that each atom only interacts with its nearest neighbor, and in a 1-D system each atom would have exactly two. So, having two interactions per atom makes sense.
There are some consequences to this type of system, which are neat but not exclusive to the model:
First, that opposite spins cost more energy. If we have a system with every spin aligned (i.e. all spins are either +1 or -1), then switching one atom to -1 will “cost” 2J energy, since it interacts with the two atoms around it.
The effect of this is that the system will tend towards a uniform spin over time, since alignment is always a lower energy state than nonalignment.
The second consequence is that this type of Ising model system will tend to become ferromagnetic, since every spin wants to be the same. However, while it’s easy to see this trend in the theoretical model, in real life, as you might expect, systems tend to be a lot more complicated, with less regular, predictable structures and neighbors, as well as a heaping of outside sources of energy (predominantly heat).
But that’s okay! It’s not what the model is for. It’s useful as an ideal model, and as a demonstration for why systems become (and remain) ferromagnetic if they are arranged in such a way as to make the lowest energy state be where their spins are relatively aligned.
Note: this should also help demonstrate why magnets have Curie temperatures, a temperature point past which they no longer are magnetic; it’s because there is a point where the energy provided by heat exceeds the associated energy cost of nonalignment.
More Ising Math
Let’s calculate the probability of any given state in the 1-D Ising model.
The amount of possible states should be obvious. Since each of the N atoms can be in two possible states, the number of configurations is 2^N. If every state were equally likely, our answer would trivially be 1/2^N for all states.
But the probability of a given state is actually more complicated. Remember that it tends to want to be more aligned, since that is the lowest energy state. So, the actual configuration probability of a state should be some function incorporating the energy—it’s Hamiltonian.
It turns out to be this guy:
Where beta is the reciprocal of the temperature and Z is just a normalization constant (to ensure the probabilities sum to 1) given by:
The solution for the one-dimensional case follows from these calculations and the Hamiltonian. So far, physicists have solved the two-dimensional case, but the three-dimensional Ising model is an unsolved problem! Pretty surprising for an ideal system.
It seems like a question with an obvious answer, but perhaps obvious in the vein of answers to questions like “What makes water wet?” or “Why is the sky blue?” There’s no quick and easy explanations, and in many cases parents who find their children asking them will discover it’s easier to say that there is no reason; they just are.
However, rubber bands actually have a very good reason for being stretchy, and the full explanation relies on thermodynamics. So worry not, future physicist parents: studying thermodynamics will let you properly answer at least this tricky question, and you will never in your life have to let another child down by explaining to them, “That’s just how rubber bands work, sweetie.” Unless you’re a lazy parent—that’s on you.
The modern rubber band is made out of natural rubber, a polymer derived from the latex of a rubber tree (synthetic rubber is generally not as stretchy). Polymers are essentially long, chainlike molecules composed of many identical or nearly identical subunits. Many polymers can sort of combine through a process called “cross-linking,” and they end up behaving in many ways more like a single molecule than a large group of them.
The cross-linked polymers of a rubber band begin in a chaotic, low energy, tangled state. When the bands are stretched, the energy is increased and the polymers untangle until they reach a local maximum of their length, and a local minimum of entropy. When released, the entropy rapidly increases until they tangle again, compressing the rubber band back to its original state.
The “cross-linking” property of polymers is vital to the band’s elasticity. Without this property, the rubber band would have no reason to tend towards a tangled state, since the entropy would be about the same in both states. If you released a rubber band with properties like that, it would just stay in the outstretched position until you force into another state.
So that’s it, right? Case closed? Rubber bands compress because of entropy; it’s beautiful, it’s elegant, and it’s eye-opening, yes, but aren’t we done?
No, silly. We have maths to do.
Rubber Band Math
An ideal “band-like” model can be constructed using a finite series of linked segments of length a, each having two possible states of “up” or “down.” For fun, let’s say one end is attached to the ceiling, and the other end is attached to an object of mass m. The segments themselves are weightless.
The entropy can be found by computing the microstates using combinatorics:
Given that any segment can be found either parallel or antiparallel to the vertical direction, the amount of segments pointing up and pointing down can be determined from N using:
and
which gives
The Boltzmann entropy is thus given by:
Applying the earlier equations:
And we get something useful!
Some follow up questions:
Given the internal energy equation U = TS + W (where W is work), find U.
Find the chain length L as a function of T, U, and given where is the tension (in this case equivalent to the gravitaitonal force mg)
Does the chain obey Hooke’s law? If so, what is the value of the stiffness constant?
Thermostatistics lies at the intersection of thermodynamics and statistical mechanics. Thermodynamics is the study of the movement of heat and energy and the heat and energy of movement. On the other hand, statistical mechanics is a branch of theoretical physics that applies principles of probability theory to study the average behavior of a system when it would be difficult to apply more direct methods (as is often the case in thermodynamics).
Statistical mechanics is kind of remarkable when you consider how people use its conceptual framework—averaging the useful properties of complex systems—on the daily. Here’s an example paraphrased from my study book: imagine going to the drug store to purchase a liter of isopropyl alcohol. For the situation at hand, this simple, volumetric specification is pragmatically sufficient. Yet, at the atomic level, we have actually specified very little.
The container which you actually want is one filled with some 8 septillion molecules of CH₃CHOHCH₃. To completely characterize the system in the mathematical formalism, you would need the exact coordinates and velocities of every atom in the system, as well as a menagerie of variables describing the bonds, internal states, and energies of each—altogether at least in the order of 1025 numbers to completely describe that thing you were able to specify earlier by just asking for “a liter” of alcohol!
Yet somehow, among all those 1025 coordinates and velocities and energies and state variables, every single one, save for a few, is totally irrelevant to describe the macroscopic system. The few that emerge as relevant are what we refer to as macroscopic coordinates or thermodynamic coordinates.
The key to this macroscopic simplicity is threefold:
Macroscopic measurements are extremely slow at the atomic scale of time.
Macroscopic measurements are extremely coarse at the atomic scale of distance.
The scope of macroscopic measurement is just about the scope of what is useful to human beings doing normal human things.
For example, to determine the size of an object too far from you to measure directly, you might take a photograph of the object with a reference scale. The speed at which this measurement takes place is determined by your camera’s shutter speed, an action in the order of hundredths of a second. On the other hand, the kinetic motion and vibration of particles at the surfaces of the object, which are constantly at work altering its observable size, act in the order of 10-15 seconds.
Macroscopic observation can never respond to such minute action. At best, under ideal circumstances, we can consistently detect macroscopic quantities of microprocesses in the range of 10-7 seconds. As such, only those combinations of coordinates that are relatively time-independent are macroscopically useful.
The word “relatively” is an important qualifier here. While we can measure processes in time quite “finely” relative to discernable human experience, it is still far from the atomic scale of 10-15 seconds.
It seems rational, then, to construct a theory to describe all the relationships of the time-independent phenomena of macroscopic systems. Such a theory is called thermodynamics.
In considering the few coordinates that are time-independent, some obvious candidates arise: quantities constrained by the conservation laws, like total energy or angular momentum, are clearly properties that are unaffected by time.
We’ll soon find that there are many more relatively time-independent coordinates dealt with in the broad scope of thermodynamics and thermostatistics.
The Thermodynamic Definition of Heat
Of the ludicrous amount of atomic coordinates, we’ve found that only a few combinations with some unique symmetry properties can survive the merciless averaging associated with transitioning to a macroscopic description. Some “surviving” coordinates prescribe mechanical properties, like volume or elasticity. Others are electrical, like the electric and magnetic dipole moments, various multipole moments, etc. Under this description, we can rewrite broad areas of physics according to which macroscopic coordinates they focus on.
Classical mechanics is then the study of one closely related set of surviving atomic coordinates. Electromagnetism is the study of another set of surviving coordinates. Thermodynamics and thermostatistics, on the other hand, are concerned with those numerous atomic coordinates that, by virtue of the coarseness of macroscopic measurement, are not defined explicitly in the macroscopic description of a system.
To illustrate, one of the most evident consequences of these “hidden” coordinates is found in energy. Energy transferred mechanically (i.e. associated with a mechanical macroscopic coordinate) is called “mechanical work,” and its macroscopic consequences are extensively treated in other areas of physics. Energy that is transferred electrically is called “electrical work,” and so on and so forth.
Notice however, that it’s just as possible for energy to transfer through the motions hidden from macroscopic measurement compared to the ones that are easily observable. Energy transfer that occurs through these hidden modes is called heat.
(Of course, this definition serves only to aid with situating heat within the macroscopic coordinate framework. We’ll soon get a more adequate working definition—basically, a mathematically sound one—to use in our studies)
So what do you say? Are we ready now for some calculations?
Like every field of physics worth its salt, thermodynamics has, at its heart, some fundamental, unchanging principles dubbed “laws.” Thermodynamics has four laws, arguably five. Inexplicably, you’ll find in the literature that they are usually not numbered 1-4 but instead 0-3. There’s probably a legitimate reason for this that I’m not bothering to find, but in my defense, how legitimate can a reason really be if it completely defies proper numbering conventions and basic logic?
No matter. The four laws, in order from, erm… zero to three, are simply stated as follows:
The Zeroth (…why?) Law of Thermodynamics – If two thermodynamic systems are each in thermal equilibrium with a third, then they are in thermal equilibrium with each other.
The First Law of Thermodynamics – Energy can neither be created nor destroyed. It can only change forms.
The Second Law of Thermodynamics – It is impossible for a process to have as its sole result the transfer of heat from a cooler body to a hotter one.
The Third Law of Thermodynamics – As temperature approaches absolute zero, the entropy of a system approaches a constant minimum.
The reason why I noted that there are arguably five laws (except, in this blasted numbering system, it would henceforth be labeled as the “fourth”) is that the all-important ideal gas law isn’t included here. Sure, it isn’t as directly related to energy transfer as the other four, but you’ll soon find that PV=nRT is involved in much more than the trivial algebra of your CHEM 101 course.
Before we go into more detail about the laws and their implications, we need to discuss what is possibly the most important yet nebulous and oft-misunderstood concept in thermodynamics: entropy.
Entropy – A Better Description
You will often hear entropy described as “disorder,” but this description is actually rather misleading; between, say, a cup that’s filled with a bunch of crushed ice cubes and a tall glass of water, the cup of water actually has the higher entropy in the context of its environment, even though you’d be hard pressed to find anyone who would argue it more “disordered.”
Basically, the issue lies in the fact that “disorder” is subjective and does not have a rigorous scientific definition, while entropy, the thing you’re trying to describe it with, does.
So there are numerous more accurate descriptions of entropy available than simply calling it “disorder” and being done with it. In my opinion, the best and most useful description for understanding thermodynamics goes like this: entropy is, at its core, a statistic. It measures the distribution of energy in a system. In particular, it measures the locations that the energy is stored in, quantifying how spread out these locations are.
At the microscopic level, energy is quantized, meaning it is measured in discrete numbers. This means that microscopic energy isn’t continuous like the real number scale, but more analogous to the list of all integers (this also means that dividing energy into “units” as we’re doing is a lot more accurate than you probably just gave me credit for).
We can effectively demonstrate this description of entropy with a simple case study. First, imagine a closed system containing two identical molecules, A and B, that each can store energy. Now suppose that each molecule has 6 distinct locations that all can store an arbitrary number of discrete units of energy, and that there are 8 energy units in total available in the system.
We can now easily see that there are many different possible states the system can take on (e.g. 1 unit somewhere in molecule A and 7 in molecule B). These are called microstates.
Let’s also assume that each distinct microstate is equally likely to occur (e.g. the state which corresponds to 2 units placed in some arrangement in molecule A and 6 in molecule B is just as likely as 4 somewhere in each), and that a microstate counts as distinct if at least one energy unit is placed in a different location.
For example, 2 energy units might be placed in the same spot in molecule A, and so both are taking up one of any of the six possible locations, or each could be in a different spot; every possible configuration of these, multiplied by all the different ways to arrange the remaining 6 energy units in molecule B, would be considered a distinct microstate.
It turns out there are 75582 ways to organize those 8 units of energy in the system we have just described. The distribution works out like this:
Energy Units in A
Energy Units in B
Possible Microstates
Probability
0
8
1287
2%
1
7
4752
6%
2
6
9702
13%
3
5
14112
19%
4
4
15876
21%
5
3
14112
19%
6
2
9702
13%
7
1
4752
6%
8
0
1287
2%
(Caution: the probabilities will questionably add up to 101%, an artifact of rounding).
If every microstate is indeed equally likely, then we can clearly see now which microstates the system will tend towards over time. The states where the energy is more evenly distributed (3 and 5, 4 and 4, etc.) are much more likely to occur than the states at the edges, where the energy is mostly concentrated in one molecule or the other. Entropy quantifies this spread of energy, and since 4 and 4 is more evenly spread than 1 and 7, it would thus have a higher entropy.
It’s important to note that entropy is proportional to the number of possible microstates in any given state. Since there are less ways to arrange the energy units when all 8 are in one molecule than when there are 4 in each, it has a correspondingly lower entropy.
This correlation between possible microstates and entropy implies that higher entropy states are more likely. If we let this system run, after a sufficient interval of time, there would be a 21% chance you will find it with 4 units of energy in each molecule, and a lower chance as you go out.
Interestingly, you may notice that there are cases, though less likely, where the entropy actually goes down. In other words, a case where the energy becomes less spread out. If we start with 5 energy units in one molecule and another with 3, there’s actually a 13% chance that the next time we check on the system, the distribution will become 6 and 2, respectively. In fact, there’s even a 2% chance that all the energy units move to the molecule with initially more energy. If this energy were stored in part kinetically (i.e. temperature, at the microscopic scale) the “hotter” molecule will have just become hotter and the “colder” molecule colder, even though they are completely free to transfer energy between each other!
This is completely counterintuitive to anyone in real life who has ever burned themselves on a hot stove. Hot objects (the pot) will always transfer energy to colder objects (your now burnt hand). Yet, in the system we just described, there’s a 21% total chance it ends up in any of the less entropic states. Certainly you’ve never touched a hot pot only to find that your hand cooled down, right? Ice cubes melt in water until they reach an equilibrium temperature, rooms get messy without deliberate intervention, and hands touching hot pots will be burned. It’s just how things work; energy wants to equalize. What gives?
To put it simply, at a macroscopic scale (the general size range of objects like burnt hands and ice cubes), the amount of atoms—and thus, places where energy can be stored—is so unimaginably high that, when you do the math, the disparity between the higher probabilities of “spread out” states and lower probabilities of states where most or all the energy is concentrated in one location is way too large for it to ever realistically be the case where, for example, your hand cools down upon touching a hot pot.
Let’s go back to our original system of two molecules. Imagine multiplying all the parameters one-thousand fold (still far from the realm of macroscopic systems), so that we now have 6000 distinct locations to store energy in each molecule (now probably more appropriately called an “object”) and 8000 discrete energy units. Now, let’s pose the obvious follow-up question.
Question: If we start from a reasonably uneven microstate—one where 6000 of the energy units are stored somewhere in object A and 2000 in object B—what’s the probability of this microstate evolving such that net energy moves from object B to A instead of the reverse, as we’d expect?
Answer: Just about 0.000000000000000000000000000003%
That’s the beauty of combinatorics: when you mess with big numbers, you’ll quickly find some ridiculous scaling.
Remember that the system which gave us that astronomically low probability is still much, much smaller than a macroscopic system. For reference, your hand is about 0.5% of your body weight, which itself has about 7 billion billion billion atoms. So, we find that a human hand is about 35,000,000,000,000,000,000,000,000 atoms.
Triple that number to estimate the amount of discrete locations where energy can be stored in it, and we can soon see that, compared to the measly 6000 locations which already gave an astronomically low chance of entropy decreasing, the probability that a system of macroscopic objects acts against the second law of thermodynamics, even briefly, is essentially too unlikely to ever occur.
Well, isn’t that remarkable? Heat doesn’t want to transfer to colder objects, as per our original intuition. The simple fact is that energy occupies whatever state it wants to. Its just that, at macroscopic scales, the higher entropy states happen to be overwhelmingly more likely than the lower entropy states by virtue of having more possible microstates. This is where the “disorder” description of entropy comes from; there are simply more ways to be disorderly than orderly. Your hand got burnt because the heat energy of the pan was allowed to evolve in its state by your touching it, and it ended up randomly (as it basically always will) with a higher entropy state of a hotter hand and a slightly colder pan.
Some additional notes: entropy can be quantified, and its quantity is oft-used in thermodynamics calculations. To give you a taste, the formula most commonly given for entropy is Boltzmann’s equation:
S = k ln(W)
where k is the Boltzmann constant equal to 1.38065 x 10^(-23) J/K and W is the number of real microstates.
Back to the Laws
Do you remember the second law? If not, here’s a refresher:
The Second Law of Thermodynamics – It is impossible for a process to have as its sole result the transfer of heat from a cooler body to a hotter one.
Did you catch the error? We know now that this statement is not entirely true. Energy transfers can sometimes occur from cooler bodies to hotter ones in a closed system, provided the system is small enough for it to be reasonably likely.
In macroscopic systems, like a car engine, your hand, or the entire universe, it may as well be true, but let’s now rephrase it to be more precise, using our newfound knowledge of entropy.
The Second Law of Thermodynamics – The entropy of an isolated system not in equilibrium will tend to increase over time, approaching a maximum value at equilibrium.
With that sorted, let’s revisit the implications of the other laws, starting with the first, er… zeroth.
The Zeroth Law of Thermodynamics – If two thermodynamic systems are each in thermal equilibrium with a third, then they are in thermal equilibrium with each other.
Not much to add here. It’s the thermodynamics equivalent of the transitive property—a=c and b=c implies a=b. Though keep in mind what it says about what you can deduce about systems in thermal equilibrium (states that are spatially and temporally uniform in temperature).
Two systems are said to be in thermal equilibrium if they are connected by a path permeable to heat and no heat transfer occurs over time.
The First Law of Thermodynamics – Energy can neither be created nor destroyed. It can only change forms.
So in thermodynamics, you’ll find that the concept of thermal energy and its relation to work is used a lot. Let’s just add a little extra onto this law to acknowledge that fact.
The First Law of Thermodynamics – Energy can neither be created nor destroyed. It can only change forms. The change in the energy of a system is the amount of net energy added to the system minus the net energy spent doing work.
Perfect.
(This can be mathematically represented as U=Q-W, where U is the total change in energy, Q is the heat energy, and W is the energy spent doing work.)
The Third Law of Thermodynamics – As temperature approaches absolute zero, the entropy of a system approaches a constant minimum.
Okay, so there are some interesting implications to this.
First, that it is impossible to reduce any system to absolute zero in a finite series of operations, which basically means that absolute zero cannot be achieved.
This also means that a perfectly efficient engine, which delivers energy in work precisely equivalent to the heat energy put in, cannot be constructed. That’s because the efficiency of a heat engine is reliant on the ratio of the difference in absolute temperature of the working hot and cold sections to the hot section (i.e. unless the cold section has an absolute temperature measuring zero, the ratio, and thus the efficiency, will be less than one).
…
That’s all for the fundamental laws, and for this section.
Interesting stuff, no? Maybe I was wrong earlier when I said thermodynamics was boring.
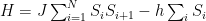
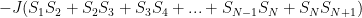


 linked segments of length a, each having two possible states of “up” or “down.” For fun, let’s say one end is attached to the ceiling, and the other end is attached to an object of mass m. The segments themselves are weightless.
linked segments of length a, each having two possible states of “up” or “down.” For fun, let’s say one end is attached to the ceiling, and the other end is attached to an object of mass m. The segments themselves are weightless.
 pointing up and
pointing up and  pointing down can be determined from N using:
pointing down can be determined from N using:



![S = k_bln(\Omega) = k_b[ln(\widetilde{N}!)-ln(N_{up}!)-ln(N_{dn})!]](https://s0.wp.com/latex.php?latex=S+%3D+k_bln%28%5COmega%29+%3D+k_b%5Bln%28%5Cwidetilde%7BN%7D%21%29-ln%28N_%7Bup%7D%21%29-ln%28N_%7Bdn%7D%29%21%5D&bg=ffffff&fg=000&s=0&c=20201002)
![S = k_bln(\Omega) = k_b[ln(\widetilde{N}!)-ln(\frac{1}{2}(\widetilde{N}-\frac{L}{a})!)-ln(\frac{1}{2}(\widetilde{N}+\frac{L}{a}))!]](https://s0.wp.com/latex.php?latex=S+%3D+k_bln%28%5COmega%29+%3D+k_b%5Bln%28%5Cwidetilde%7BN%7D%21%29-ln%28%5Cfrac%7B1%7D%7B2%7D%28%5Cwidetilde%7BN%7D-%5Cfrac%7BL%7D%7Ba%7D%29%21%29-ln%28%5Cfrac%7B1%7D%7B2%7D%28%5Cwidetilde%7BN%7D%2B%5Cfrac%7BL%7D%7Ba%7D%29%29%21%5D&bg=ffffff&fg=000&s=0&c=20201002)
 where
where  is the tension (in this case equivalent to the gravitaitonal force mg)
is the tension (in this case equivalent to the gravitaitonal force mg)